
 Home
HomeInquiries on the study
Hidemasa Bono
Professor, Graduate School of Integrated Sciences for Life
E-mail: bonohu * hiroshima-u.ac.jp
Kouhei Toga
Researcher, Graduate School of Integrated Sciences for Life
E-mail: togchemi * hiroshima-u.ac.jp
(Note: Please replace * with @)
Inquiries on the story
Hiroshima University Public Relations Office
E-mail: koho * office.hiroshima-u.ac.jp
(Note: Please replace * with @)
Researchers have identified “DNA switches” that become active as honeybee larvae grow into worker bees, offering new insight into the development of these important pollinators and the ecosystems they support.

Honeybee (Apis mellifera). Credit: Ivar Leidus, CC BY-SA 4.0, via Wikimedia Commons
Caterpillars turning into butterflies. Tadpoles transforming into frogs. Grubs metamorphosing into bees, beetles, wasps and ants.
Metamorphosis is the remarkable biological process by which some species of animals and insects physically transform into adults through sequential, and very different, developmental stages. In complete metamorphosis, roughly 75–80% of insect species transition through four different life cycles—egg, larva, pupa and adult—before transforming into mature adults.
The transition between developmental stages in complete metamorphosis is particularly interesting from a genetic standpoint, as transforming insects and animals have the same genome in each developmental stage yet look and behave often very differently at larval versus adult stages. Today, researchers are beginning to understand the genetic mechanisms that regulate these complete transitions between developmental stages in metamorphosing insects and animals. Additionally, studies are revealing how insects like bees, ants, wasps and termites can produce distinct social castes—queens and workers—from genetically identical larvae by altering gene regulation.
Recently, a group of researchers from Hiroshima University used cap analysis of gene expression (CAGE) technology to assess the activity of computationally predicted honeybee enhancer sequences that could increase the expression of nearby genes during worker bee metamorphosis. Enhancer sequences are regions of DNA that work like switches or volume controls, helping regulate when certain genes turn on and how strongly they act. Notably, the study provides the first direct evidence of enhancer sequence activity during honeybee worker metamorphosis using CAGE technology.
The team published their study on May 19, 2026 in the journal Insects.
“Our study asks which enhancers are actually active during honeybee (Apis mellifera) worker metamorphosis and which transcription factors use them to regulate key developmental genes. This matters because a previous study predicted transcription factor binding sites computationally from genome sequence alone, and direct evidence of activated enhancers across sequential developmental stages in worker bees has been lacking,” said Hidemasa Bono, professor in the Graduate School of Integrated Sciences for Life at Hiroshima University in Hiroshima, Japan.
Specifically, the team sequenced the very beginnings of worker bee mRNA molecules and mapped those sequences back to the honeybee genome to identify transcription factor (TF) binding sites (BSs). TFBSs are locations in the genome where gene expression proteins assemble on DNA to express specific genes. By locating TFBS, the research team identified the location of 842 potential enhancer sequences, including many intronic enhancer sequences, that can bind activator proteins and increase the likelihood of gene expression.
Importantly, this study identified enhancer sequences based on enhancer RNA from actual worker honeybees, rather than simply predicting enhancer sequences based on genome sequence alone.
“Changes in gene expression levels can be readily identified using transcriptomic analysis. However, the regulatory transcription factors driving these changes remain largely unidentified, because most TFBSs within enhancers are inferred from sequence-based conservation rather than direct observation of activity. Providing experimental evidence of active enhancers is therefore valuable for understanding the evolution of the highly sophisticated sociality seen in honeybee Apis mellifera,” said Kouhei Toga, researcher in the Graduate School of Integrated Sciences for Life at Hiroshima University.
The researchers classified the 17,349 transcription start sites (TSSs) and 842 candidate enhancers into five categories based on their overall expression patterns. These clusters were regulated by transcription factors cycle and vismay, ttk, ovo and paired, GATAe, and daughterless in clusters one through five, respectively. From these categories, the team further narrowed worker honeybee regulatory relationships to just 15 specific transcription factor–enhancer–target gene relationships controlling metamorphosis. Importantly, genes associated with known metamorphic regulators Broad complex (Br-c) and E93 were found within specific clusters.
During their analysis, the research team found transcription factor tramtrack (ttk) binding sites in five honeybee enhancers associated with four target genes, including Br-c. Surprisingly, ttk-binding sequences within these enhancers are perfectly conserved across the genus Apis, yet differ from those found in other bee lineages, such as bumblebees. This single-nucleotide difference suggests that honeybees may have acquired a unique transcriptional regulation during the evolution of their highly sophisticated social caste system—one that other bees simply do not possess.
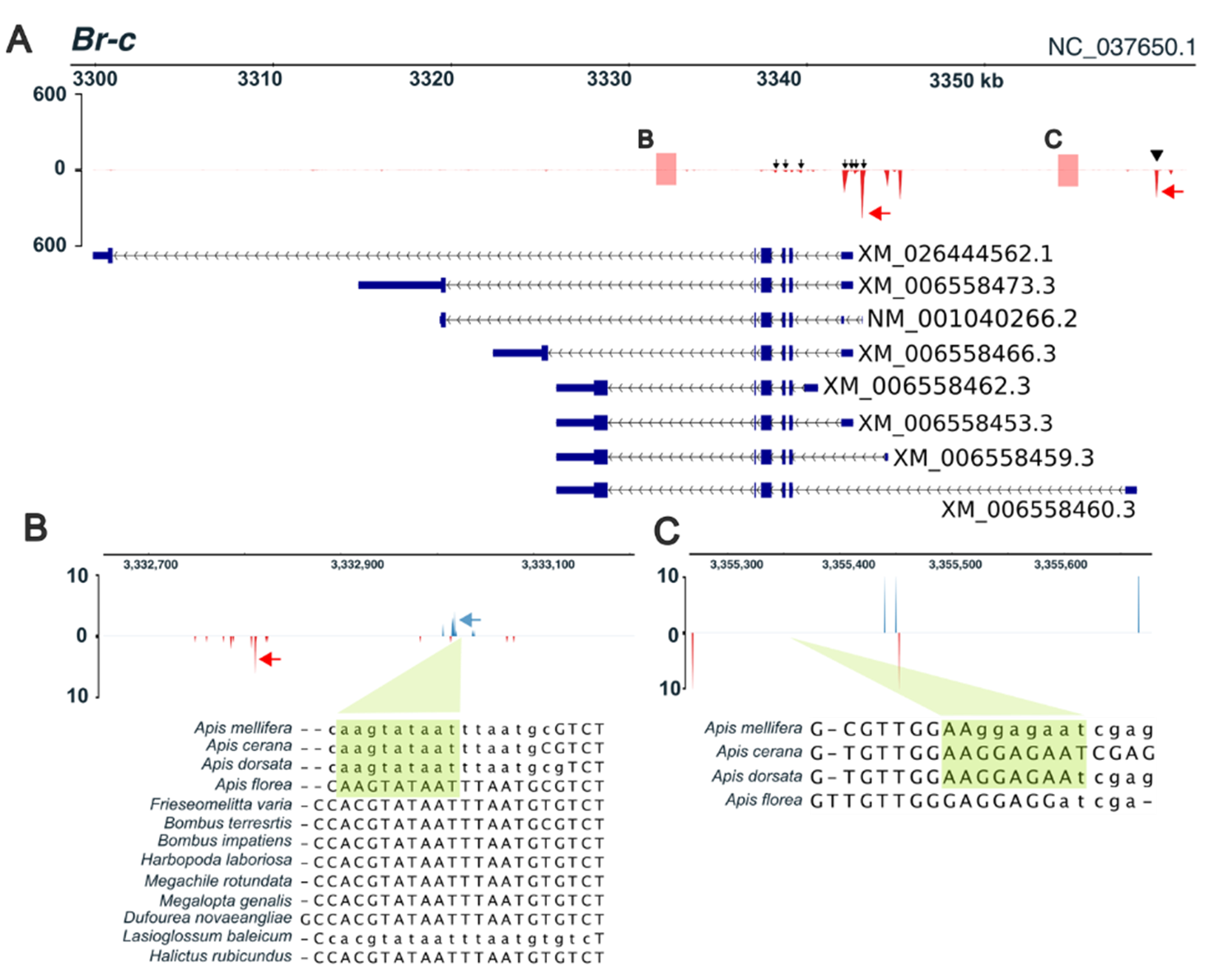
CAGE can detect RNAs transcribed in opposite orientations, allowing enhancer regions to be quantitatively identified as regions where bidirectionally transcribed RNAs are present at positions separated by several hundred base pairs or more. In all panels, red (red arrows) and blue peaks (blue arrows) indicate signals on the negative strand and positive strand, respectively. Green boxes indicate predicted ttk binding sites (derived from Drosophila melanogaster motifs). Gene models are shown in blue. Corresponding enhancer regions in other bee species are displayed at the bottom of each panel. Lowercase nucleotide sequences represent repeat regions in the genome. The vertical axis represents transcription start site count data calculated by CAGE. (A) Gene structure and transcription start sites of Br-c. Arrowheads and arrows indicate individual transcription start sites. Red boxes represent enhancer regions identified in this study; regions B and C are magnified in panels (B, C). (B) An intronic enhancer region within Br-c (magnified view of region B in panel (A)). (C) An additional intronic enhancer region within Br-c (magnified view of region C in panel (A)). Adapted from Toga et al. (2026) Insects, https://doi.org/10.3390/insects17050516. Originally published under CC BY 4.0. This version has been modified from the original.
While the application of CAGE technology confirmed the presence of worker enhancer sequences in Apis mellifera honeybees, the team still needs to validate their results using different assays to build a more comprehensive picture of the gene regulatory networks controlling worker development. Ultimately, the team would like to use this knowledge to address pollinator challenges occurring worldwide.
“Honeybees serve as primary pollinators for a wide range of crops, including strawberries, and play a critical role in maintaining biodiversity. A deeper understanding of the molecular mechanisms governing worker development therefore has far-reaching implications, not only for apiculture but also for global food security and ecosystem conservation,” said Bono.
This research was supported by the Center of Innovation for Bio-Digital Transformation (BioDX), an open innovation platform for industry-academia co-creation (COI-NEXT) of JST (JPMJPF2010), and the RIKEN-Hiroshima University Joint Research Program for Science and Technology Hub.
About the study
- Journal: Insects
- Title: Genome-Wide Identification of Transcriptional Start Sites and Candidate Enhancers Regulating Worker Metamorphosis in Apis mellifera
- Authors: Kouhei Toga, Kakeru Yokoi & Hidemasa Bono
- DOI: 10.3390/insects17050516
Media Contact













