Home
Home 広島大学大学院先進理工系科学研究科 辻 聡樹
Tel:082-424-7680
E-mail:tsuji*hiroshima-u.ac.jp
(*は半角@に置き換えてください)
本研究成果のポイント
- 創薬や材料開発における電子レベルの高精度なコンピュータ・シミュレーションの実用化に向けて、大規模な量子化学計算(*1)を可能にする計算量削減手法を10倍以上高速化しました
- 最新のGPU(*4)アーキテクチャを活用した計算量削減手法の高速化によって、100原子を超えるハートリーフォック法(*2)やポストハートリーフォック法(*3)が数秒から十数秒で実行可能になります
- 広島大学発のオープンソース量子化学ソフトウェア「GANSU(*5)」で一般に利用可能となります
- さらにGPUを用いたフラグメント分割手法によって、従来は2か月以上かかっていた300原子以上のCCSD計算[6]が1.4時間で完了しました
概要
広島大学大学院先進理工系科学研究科の伊藤靖朗教授らの研究チームは、富士通株式会社と共同で大規模な量子化学計算を可能にする計算量削減手法を大幅に高速化しました。ハートリーフォック法に必要なメモリ量を削減する「Direct-SCFアルゴリズム(*6)」に加えて、「密度フィッティング(RI近似)(*7)」や「フラグメント分割手法(*8)」などの計算量削減手法を高速化することで、GPUを用いて大規模かつ高精度なポストハートリーフォック法を短時間で実行できるようになりました。特にフラグメント分割手法を用いた大規模量子化学計算では、372原子のCCSD計算が1.4時間で完了し、計算量削減手法を使わない場合に対して1,295倍の高速化を達成しました。
今回の研究成果は、2025年3月に公開したオープンソースの量子化学ソフトウェア「GANSU」に実装され、一般に利用可能となります。また、本研究成果は2025年4月25日に学術論文誌「Applied Sciences」に掲載されました。
背景
新しい医薬品や高機能な材料を開発するプロセスでは、無数の候補化合物についてその性質を調べて最適なものを見つけ出す必要があります。この探索を、量子力学の理論に基づいた電子レベルのコンピュータ・シミュレーションを用いて高精度に実現できる量子化学計算は、実験にかかる膨大な時間とコストを削減できるためその実用化が大きく期待されています。しかし量子化学には、対象とする原子数など化合物のサイズが大きくなるほど、計算に必要な時間とメモリ量が爆発的に増加するという計算量の壁が存在します。この壁を打破するために、量子化学の中心的なフレームワークであるハートリーフォック法やポストハートリーフォック法では、様々な計算量削減手法が開発されています。
一方でそれらの手法を、専門性が異なる情報・計算機科学の知見を持って最新のコンピュータ・アーキテクチャに向けて効率的に実装するためには技術的な課題が残されています。こうした背景も一因となり、これまで量子化学計算は扱える化合物サイズの限界や計算コストの問題が解決できず、創薬や材料開発における実用化が制限されてきました。
研究成果の内容
広島大学と富士通株式会社の共同研究講座「富士通次世代コンピューティング共同研究講座」では、並列分散処理やGPUプログラミングの技術を駆使して、量子化学計算を抜本的に高速化する研究開発に取り組んでいます。今回の研究成果では、GPUを用いて量子化学における3つの計算量削減手法を高速化することで、ハートリーフォック法やポストハートリーフォック法の計算性能を実用化に向けて大きく前進しました。
ハートリーフォック法は、原子数のおよそ4乗に比例する組み合わせの電子に関する積分計算(二電子積分)とその値をメモリに保存するためのデータ使用量が大きなボトルネックとなっています。そのため100原子を超える規模では、これらの積分値がCPUやGPUのメモリ容量に収まりきらないため計算が困難になります。そこで本研究では、二電子積分を保存せず必要なときにオンザフライ計算することでメモリ使用量を原子数のおよそ2乗に削減できる「Direct-SCFアルゴリズム」に注目し、GPUを用いて並列化および高速化をしました。膨大な積分値を効率的に集約する並列アルゴリズムを提案することで、300原子以上の分子についても高速なハートリーフォック法の実行を可能にしました。今回の研究成果によって、既存の量子化学ソフトウェアに対して最大13.1倍の高速化を実現しました。
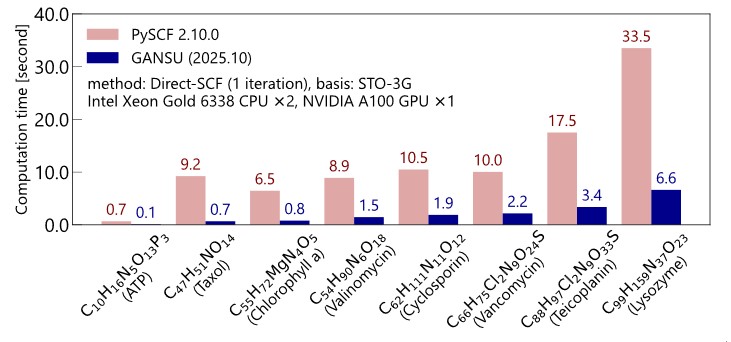
図1. Direct-SCFにおけるフォック行列の計算時間
また、二電子積分を近似して求めることで必要なメモリ量を原子数の4乗からおよそ3乗に削減できる「密度フィッティング(RI近似)」と呼ばれる高精度な近似手法についても、GPU実装および高速化を実施しました。これはハートリーフォック法だけでなくポストハートリーフォック法においても有効な近似計算手法であり、より高精度な大規模量子化学計算を実現することができます。GANSUでは密度フィッティングを用いてポストハートリーフォック法のひとつであるメラープレセット法が実装され、およそ200原子規模の分子において既存の量子化学ソフトウェアに対して最大12.7倍の高速化を達成しました。
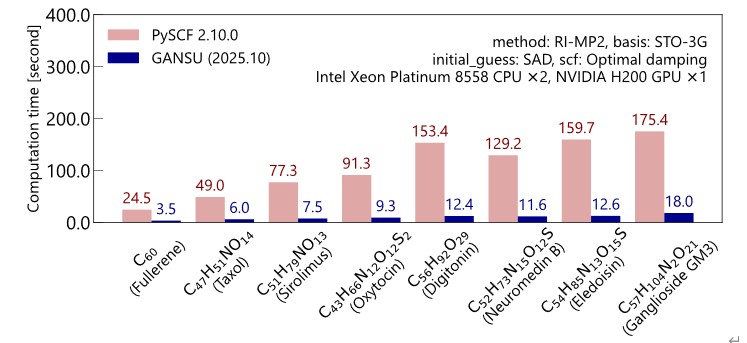
図2. 密度フィッティングを用いた二次のメラープレセット法(RI-MP2)の計算時間
しかし創薬や材料開発で取り扱われるより大きな規模の化合物について高精度なポストハートリーフォック法を実現するためには、原子数の5乗や6乗といったさらに爆発的に増加する計算量の壁を打破する必要があります。これを解決するアプローチとして、大規模な化合物をフラグメントと呼ばれる小さな部分系に分割することで計算量を大幅に削減する「フラグメント分割手法」の高速化に取り組んでいます。富士通次世代コンピューティング共同研究講座では、フラグメント分割手法のひとつであるDMET (Density Matrix Embedding Theory) というアルゴリズムを高速化しました。分割された各フラグメントに対してひとつのGPUを割り当て、さらにマルチGPUによって複数フラグメントの計算を並列に実行することで高速なポストハートリーフォック法を実現しています。またこの手法は、化合物を分割して計算することによる近似誤差が発生するため、この精度誤差を最小化する分割方法が極めて重要です。そこで富士通株式会社より研究開発されている自動分割手法[5]を活用し、計算精度を維持できるようにフラグメントを作成しています。GPUクラスタを用いた今回の大規模実行では、創薬の分野で対象となるタンパク質と薬剤候補化合物の結合部分に含まれる372原子について、DMETを用いた結合クラスタ法(CCSD)を1.4時間で完了しました。これにより従来のCCSD計算にかかる見積もり1,813時間[6]に対して1,295倍の高速化を達成しました。
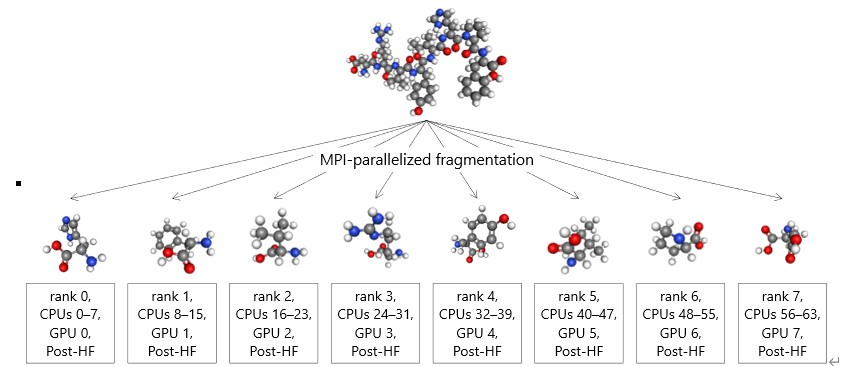
図3. フラグメント分割手法DMETのマルチGPU実行
これらの計算量削減手法を大幅に高速化した今回の研究成果は、高精度な量子化学計算によって創薬・材料分野の開発プロセスを大幅に加速する基盤技術となります。電子レベルの高速なエネルギー計算や分子構造の最適化を実現することで、薬剤などの候補化合物を効率的に探索できるだけでなく、それらを予測・生成するAIの高精度な学習データを大量に作成することも可能となります。
今後の展開
これらの計算量削減手法をオープンソースの量子化学ソフトウェアGANSUに実装および順次公開することで、大規模な量子化学計算が一般に利用可能となります。今後は1,000原子などさらに大規模かつ高速なポストハートリーフォック法の実現に向けてスーパーコンピュータを活用した分散並列実装などを目指します。
論文情報
論文情報
[1] タイトル:GPU-Accelerated Fock Matrix Computation with Efficient Reduction
掲載誌:Applied Sciences
著者:Satoki Tsuji, Yasuaki Ito, Haruto Fujii, Nobuya Yokogawa, Kanta Suzuki, Koji Nakano, Victor Parque, and Akihiko Kasagi
責任著者:Satoki Tsuji
DOI:https://doi.org/10.3390/app15094779
掲載日:2025年4月25日
関連論文
[2] Dynamic Screening of Two-Electron Repulsion Integrals in GPU Parallelization, CANDAR 2024 workshops, pp. 211–217.
https://doi.org/10.1109/CANDARW64572.2024.00041
[3] Efficient GPU Implementations of Three-Center Two-Electron Repulsion Integrals, Concurrency and Computation: Practice and Experience, vol. 37, no. 25-26, 2025.
https://doi.org/10.1002/cpe.70328
[4] GPU Acceleration of RI-RMP2 Correlation Energy Computation, CANDAR 2025, to appear.
[5] Automatic Molecule Fragmentation for Density Matrix Embedding Theory, The Journal of Physical Chemistry A 2025 129 (40), 9511-9520.
https://doi.org/10.1021/acs.jpca.5c06027
[6] Accurate and Fast Geometry Optimization with Time Estimation and Method Switching, arXiv preprint arXiv:2404.12842, 2024.
https://doi.org/10.48550/arXiv.2404.12842
用語解説
(*1)量子化学計算
分子のエネルギーや構造を量子力学の理論に基づいて高精度に計算する手法
(*2)ハートリーフォック法
個々の電子による影響を平均化して取り扱う量子化学の基本的な近似計算手法
(*3)ポストハートリーフォック法
ハートリーフォック法では考慮できない電子相関を取り入れてさらに高精度な計算を実現する手法の総称
(*4)GPU (Graphics Processing Unit)
科学技術計算や機械学習などにも用いられる並列計算性能が高いグラフィックス処理に適した専用プロセッサ
(*5)GANSU (GPU Accelerated Numerical Simulation Utility)
GPUを用いて高速な量子化学計算を実現する広島大学発のオープンソースソフトウェア
https://github.com/Yasuaki-Ito/GANSU
(*6)Direct-SCFアルゴリズム
二電子積分を保存せず必要なときにオンザフライ計算することでメモリ使用量を原子数の4乗からおよそ2乗に削減できる手法
(*7)密度フィッティング(RI近似)
二電子積分を近似して求めることで必要なメモリ量を原子数の4乗からおよそ3乗に削減できる手法
(*8)フラグメント分割手法
大規模な化合物をフラグメントと呼ばれる小さな部分系に分割することで計算量を大幅に削減する手法
【お問い合わせ先】